Transmembrane Proteins

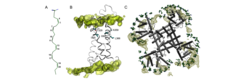
The voltage-gated channel protein Shaker is a tetrameric, integral membrane protein that forms aqueous pores through which K+ can flow across nerve membranes. Polyunsaturated fatty acids (PUFAs) are natural components of the lipid bilayer in e.g. heart and nerve cells and appear to have a significant effect on and modulate voltage-gated K+ channels and are used for example in dietary treatments of epilepsy. The effects of PUFA-channel interactions have been postulated to involve opening of K+ channels, but the molecular mechanism remains unknown. Molecular dynamics simulations of the Shaker K+ channel embedded into a PUFA-enriched membrane proposed specific molecular PUFA-channel interactions. The observed packing environment differed in the open and closed states of the channel protein.
G protein-coupled receptors (GPCRs) constitute the largest family of cell-surface receptors that are involved in cell signal transduction pathways and regulate numerous cellular processes in human tissue. Although all GPCRs share a common feature of a 7-transmembrane (TM) spanning domain, there are still many challenges in terms of crystal determination since the protein is embedded within the lipid bilayer. The dopamine D2 receptor (D2R) and its structural role as a drug target was investigated in detail and stands as one example of the GPCR family. Small molecule that bind D2R are drugs to treat Parkinson's disease (PD), various psychotic disorders such as schizophrenia, and particularly the common prolactinomas of the anterior pituitary gland.


Binding of the most common antagonistic schizophrenia drugs (clozapine, olanzapine, risperidone, quetiapine, and ziprasidone) was simulated in silico. Their binding modes to a comparative D2R model based on the β-2-andronergic receptor were investigated in detail and binding selectivities to different receptor conformations could be elucidated and are in excellent agreement with experiment. With the availability of highly efficient quantum mechanical, (here DFT) calculations, QM calculations of these drug molecules to large cluster models of 200-300 atoms were performed. The QM-calculated ligand binding affinities for MD-refined poses were significantly lower (by −15 to −8 kcal/mol) than docking scores and in a much better agreement with experimental binding energies.